

Abstract
Introduction: It is widely accepted that primary Hemophagocytic Lymphohistiocytosis (PHLH) in infants and children is a genetic disease inherited in an autosomal recessive manner. However, it is proposed recently that the late-onset PHLH could result from the synergetic effects of the atypical heterozygous HLH-associated mutations and trigger of environmental factors such as viral infection. EBV is the most common viral pathogen linked to the development of HLH. EBV is known as a trigger to induce secondary HLH in some patients; it also can be a trigger of PHLH. However, the subpopulation of EBV infection in PHLH is unknown. Identification of EBV-infected lymphocyte subsets will help to clarify the role of EBV in the development of late-onset PHLH. This study identified the EBV infected cells subpopulation and summarized the clinical and genetic characteristics of 10 late-onset PHLH patients.
Methods: Between June 2013 and June 2018, 10 patients with Epstein-Barr virus-triggered Late-onset PHLH and their family members were investigated, with a median age of 22.3 years (range, 12-45 years). We quantified EBV load by PCR, examined Natural killer (NK) cell activity by Lactate dehydrogenase (LDH) assay, and performed flow cytometry to analyze perforin expression and degranulation in addition to collecting clinical characteristics of 10 EBV-triggered PHLH patients. Next-generation sequencing was used to detect HLH-associated genes mutations. Immunobead sorting followed by quantitative PCR and fluorescence in situ hybridization were conducted to identify EBV-infected cells. We also performed pedigrees investigation and detected EBV loads and HLH-associated genes mutations in these patients' family members.
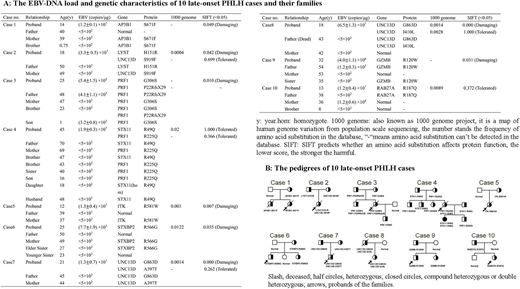
Results: All patients had the acute onset and rapid progression of a disease and met the Diagnostic Guidelines for HLH (2004) proposed by the Histiocyte Society. Reduced NK-cell activity was observed in 6 late-onset PHLH patients (normal range: 30%-50%), while NK-cell activity defect (<5%) was found in 4 patients. Substantially reduced perforin expression (normal range: > 50%) on CD3-CD56+ NK cells were detected in 7 of 10 patients (70%). One of 10 cases (10%) had abnormal resting NK-cell degranulation, and 4 of 10 (40%) had defective resting NK-cell degranulation. Abnormal activated NK-cell degranulation (normal range: > 20%) was recorded in 3 of 10 cases (30%). Nine of 10 cases (90%) had either reduced perforin expression or decreased activated NK-cell degranulation. The EBV-DNA load and genetic characteristics of the late-onset PHLH cases and their families were summarized in Figure A. All the details of 10 pedigrees are shown in Figure B. The family members had the same type of mutations as the patients but did not develop the disease. High load of EBV was detected in the peripheral blood mononuclear cells of the probands, whereas a much lower load of EBV were found in the probands' family members by quantitative PCR. The results of sorting-PCR demonstrated that three group of cells (T cells, B cells, and NK cells) in all the patients were infected with EBV, in 9 of them EBV significantly infected NK cells, and only in 1 patient EBV infected both NK cells and T lymphocyte cells primarily. More than half of the patients (7 of 10) died of disease progression or complications, and median survival time is only 95 days. Only three patients who received allogeneic hematopoietic stem cell transplantation from unrelated donor acquired long-term survival, currently alive.
Conclusions: The late-onset PHLH is a devastating disorder, and the prognosis is poor. Unlike sporadic HLH, EBV-associated late-onset PHLH patients usually have 1 or 2 pathologic mutations of PHLH-associated genes, reduced perforin expression or degranulation. Some of them have abnormal T or NK cells in bone marrow or lymph node; most of them have high load EBV infection, and the target cells of EBV infectious are usually NK cells, sometimes combined with T cells. HLH gene sequencing and pedigree investigation can help clinicians classify patients as having late-onset PHLH or sporadic HLH. Based on genetic mutations, EBV-Infected NK/T lymphocyte subtypes may be involved in the development of Late-onset PHLH.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal